


Présentation de l'équipe
GIMAP is a multidisciplinary research group comprising immunologists, microbiologists, infectiologists, gastro-enterologists, nephrologists... The GIMAP was created in 1991 as a group of Jean Monnet University (UJM) to initially develop a research on mucosal immunology related to genital infection by HIV. The unit is a major actor of UJM that federates a coherent and original research at cross-roads between mucosal immunology, inflammation and infection led by seventeen UJM professors. It is composed of lecturer-researchers mainly from the medical sector, scientists (including one engineer), four technicians, a secretary and students at different levels .The team is located at the Faculty of Medicine of Saint-Etienne, localized on the “Santé Innovation” Campus of the University Hospital of Saint-Etienne since 2015. Our group is part of the « Institut Fédératif de Recherche IFRESIS » (FED 4166) and relies on a Clinical Investigation Center (CIC-EC 1408), including a Vaccinology axis member of iREIVAC consortium (member of the PRESAGE institute) and on an animal experimentation platform (PLEXAN).
GIMAP would like to decipher in details the particular network of interactions between pathogens/commensal and secretory immunoglobulins. We will focus on the description of facilitating or escape mechanisms which involve SIgs and microbiota into the gut and the nasal mucosa. Finally we will also address if the fantastic ability of secretory Igs to cross all the monostratified epithelium could be used for vaccine approaches.
1) Study of the interactions between secretory Igs and gut microbiota
Based on our previous results in IBD and the putative facilitating role of SIgA to promote inflammation in IBD, we would like to extend our work in different other gut associated inflammatory situations as chronic infections with HIV or CMV but also during ageing.
First we would like to ask the key question about the ability of IgA or IgM able to promote the emergence of viral gut reservoir in two clinical situations. First, we will address the role of AMIS in the emergence or elimination of HIV gut associated reservoir in chronic infected patients but also in Long-Term Non Progressors (LTNP) and Immunological Non Responders (INR). Does IgA or IgM facilitate the HIV infection of gut CD4 activated T cells by protecting viral particles and promoting the cross-link of HIV/Igs complexes with targeted cells? Does IgA or IgM regulate the composition of micobiota during HIV infection and promote GALT inflammation necessary for HIV infection and result in strong dysbiosis? How Igs regulated dysbisosis will affect mucosal immune response against HIV and in particular the Treg/Th17 balance? These studies will be conducted both in vitro/ex vivo in patients but also modeled using preclinical experimental models as SCID-HIS HIV infected mice. We will evaluate the composition of IgA+/IgA- or IgM+/IgM- microbiota in those patient populations and correlate it to metabolomic study as the alteration of tryptophan catabolism. It will help us to have a clear picture about the facilitating role during HIV infection or in their ability to promote gut-associated inflammation. In a second project, we would like also to investigate how Igs could promote the emergence of CMV reservoir in IBD patients. We have described that the prevalence of CMV is highly increase in Ulcerative Colitis patients in contrast to Crohn patients. Ulcerative Colitis has been described to be a Th2-based inflammatory disease. We would like to evaluate if Igs could influence CMV infection in UC situation and promote or regulate the composition of microbiota. We will model thisclinical situation in a relevant colitis preclinical model with murine CMV chronic infection. We will descibe the profile of IgA or IgM responses against CMV in those patients and characterize if IgA/CMV or IgM/CMV complexes have the ability to strongly infect epithelial cells in vitro and in vivo. We will then look if IgA+ or IgM+ microbiota is altered in UC patients and defined new insights in the inflammatory process of UC.
In parallel, we would like to continue our characterization of the role of AMIS in Crohn disease (CD). We have shown that IgA promote an important translocation of pathogens in CD patients. We would to characterize in details the IgA+ and IgM+ microbiota in inflammatory and non-inflammatory patients. This characterization will be done into stool samples but also into gut biopsies with FISH labeling of 16S RNA and IgA-specific antibody. We would like to know if IgA1 or IgA2 but also the different isoforms of IgA (monomeric or dimeric) are able to promote dysbiosis and inflammation in CD patients. We will also look in vitro if isotypes or isoforms of secretory Igs (IgA&IgM) regulate differentially the mucosal immune response (activation of dendritic cells or macrophages) and if the ability of secretory Igs to regulate immunity is modified during ageing (VITAL IMI project). There is to date any datas about the effect of ageing on the functions and AMIS ability so this is an important issue. We will also try to decipher the mechanisms involve in the IgA-mediated dysbiosis mechanisms. We obtained by mass spectrometry interesting results about glycosylation and sialylation modifications of IgA1 and IgA2 in CD patients. We will pursue our characterization of IgA and IgM phenotypes in CD patients (glycosylation, sialylation, glycan reactivity by high throughput screening and specificity of mucosal B cell clones). Modification of IgA1 glycosylation pattern has been also described by our team in IgA nephropathy. To explore the gut-renal axis, relevant in IgA nephropathy pathophysiology, we aimed at developing a genetically altered mouse model comprising the expression of human IgA1 and the suppression of PIGR (Polymeric Ig Receptor). This model has been designed to induce the overrepresentation of polymeric IgA1 originating from the gut to be redirected in the systemic compartment, corresponding to a trait of IgA nephropathy. Once done, various stimulations are being explored to trigger an actual nephropathy, such as mucosal challenges of bacterial homogenates, antibiotics, fecal transplantation to induce dysbiosis and modify microbiota.
2) Study of the interactions between secretory Igs and nasal microbiota
Colonization of the nasal mucosa is a field which is not well understood. Nasal Igs+/- microbiota is not well known to date. The role of IgA or IgM in the shaping of nasal microbiota is an important question. We plan to study how some pathogens such as Staphylococcus aureus (SA) could evade AMIS during colonization. The role of SIgA in the physiopathology of SA nasal colonization is still unknown (Mulcahy et McLoughlin 2016 Trends Microbiol.). In mice, glycosylation of IgA-specific Asn-442 blocks IgA binding pathogen proteins, which react with the central CH2/CH3 interface, but still permits interaction of pIgR to preserve SIgA mediated mucosal immunity (Wines et al. J. Biol. Chem. 2011). Although it is a highly glycosylated protein, its site-specific glycosylation and associated glycan micro-heterogeneity has still not been fully elucidated. In humans, multiple glycosylation sites were identified with nearly 30 glycan compositions located at seven sites on the secretory component, six compositions at a single site on the J-chain, and 16 compositions at five sites on the IgA heavy (H) chain (Huang et al. J. Proteome Res. 2015) We plan to study how SIgA glycoforms found in humans could modify the interactions with SA proteins such as SSL7 or staphylococcal proteases and regulate SA nasal colonization as previously described for few pathogens (Chi et al., Prot. Sci. 2017; Ramsland et al., PNAS 2007; Wines et al., JBC 2011).
We have shown that Dectin-1, which is mainly expressed by M-cells, promoted the internalization and the transcytosis of both free secretory IgA (Rochereau et al. PLoS Biol. 2013) and IgA-coated Salmonella (Gayet R, PhD thesis, GIMAP 2015-2018). Because (i) SA secretes Staphylococcal Superantigen-like (SSL) 7 that bind the heavy chain of IgA at the CH2/CH3 interface (the junction between constant domains 2 and 3 of the heavy chain)(Langley et al. J. Immunol. 2005; Ramsland et al. Proc. Natl. Acad. Sci. U. S. A. 2007; Wines et al. J. Biol. Chem. 2011) and (ii) Dectin-1 could be upregulated at the surface of epithelial cells during inflammatory diseases (e.g. nasal polyposis) (Gong et al. Am. J. Otolaryngol. 2013) or infectious diseases,(Lee et al. J. Clin. Immunol. 2009) we hypothesized that SIgA could facilitate the internalization of SA in both nasal epithelial cells and M cells found in the NALT. In addition to facilitating the bacteria internalization, as described with Shigella flexneri in mice (Mikulic, Bioley, et Corthésy 2017 J. Mol. Biol.) recognition of SIgA-SA immune complexes by Dectin-1 could participate in controlling locally mucosal DC responsiveness to foster non-inflammatory cytokines and chemokines production. Once into the cell, little is known about the evolution of the endosome-associated SA. Depending on the way of SA entrance (receptor and signalling pathways), the fate of the bacteria could range from rapid clearance to persistence. Autophagy, which is a highly conserved lysosomal-dependent catalytic intracellular mechanism that participates to pathogen control in eukaryotes (namely xenophagy), has been identified as a pivotal mechanism (Horn et al. Int. J. Med. Microbiol. 2017, Faure et al. J Innate Immun. 2013). Phenol soluble modulins secreted by SA were found to promote autophagy and cytotoxicity by damaging endosomal membranes (Dupieux et al. EP0365, ECCMID 2017). We plan to unravel how SA subverts the autophagy process through the phosphorylation of p38a/MAPK14 and ATG5. Little is known about the modulation of SA- induced autophagy and notably whether autophagy fosters SA intracellular survival or clearance remains unclear at least because cell models and SA strains used differs from one study to another (Neumann et al. Autophagy 2016, Schnaith et al. J. Biol. Chem 2007). In parallel, ongoing clinical trial aimed to study pathogen nasal carriage and decolonization strategies in hospitalized patients in view of AMIS mechanisms.
3) Use of secretory Igs in mucosal-targeted biotherapy approaches
Based on the role and the properties of SIgs to cross monostratified epithelium as in the GALT or in the NALT, we developed new vaccine strategies to deliver antigens into the mucosa (Rochereau et al., Eur. J. Immunol. 2015; Rochereau et al., JACI 2016). We will continue our efforts to develop highly potent mucosal delivery platform based on the use of secretory IgA or IgM as multivalent carrier and new adjuvants able to stimulate strong mucosal immune responses by cross-linking of extracellular or intracellular PRRs for vaccine purposes but also for eradication of mucosal pathogenic reservoirs (Gutjahr et al., 2017, 2019; Pavot et al., 2014, 2015). We will evaluate the ability of our mucosal delivery platform to be used as a multivalent vaccine approach for leptospirosis (ANR project THAIVAC), Salmonella typhimurium and for viral and bacterial respiratory diseases in the veterinary field with Boehringer Ingelheim.
Alternatively, specific SIgs from human biopsies or induced in humanized SIgs mice will be also used to block mucosal transmission of pathogens as HIV (CAPRISA trials). The use of polyclonal/polyspecific IgA to block mucosal infection or to restore IgA deficiency will be also evaluated.
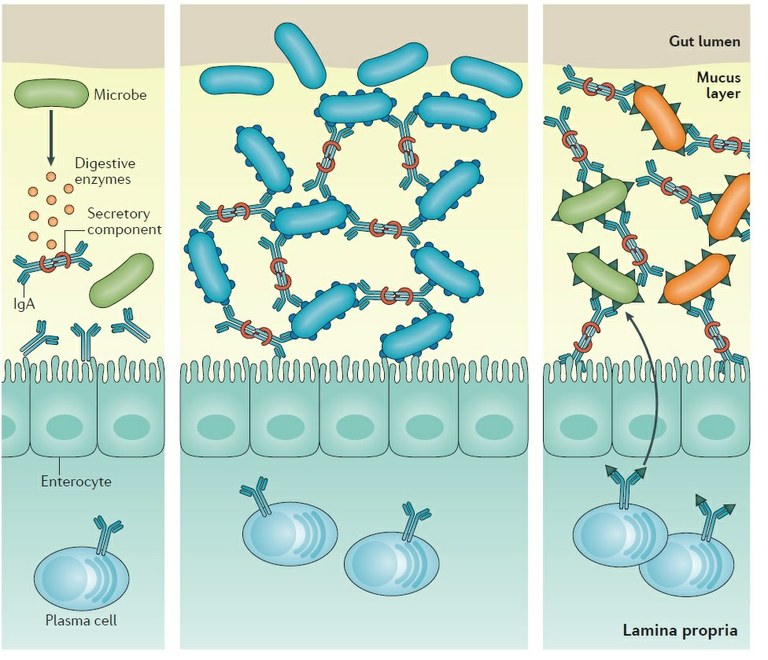
Figure 1: Role of secretory Igs in infectious and non infectious mucosal inflammation. Antibody–microorganism interactions that influence antibody-mediated immunoselection of the microbiota. Some microorganisms can secrete enzymes that degrade the secretory component of secretory antibodies, which disrupts the stability of the antibody molecule, rendering it ineffective. Some microorganisms may express surface epitopes that bind secretory IgA and facilitate colonization of the mucus lining in the gut. Some microorganisms express surface epitopes that are very similar to those on other microorganisms. Antibodies generated against one microorganism can lead to the generation of antibody specificities that cross-react with similar epitopes found on other microorganisms.
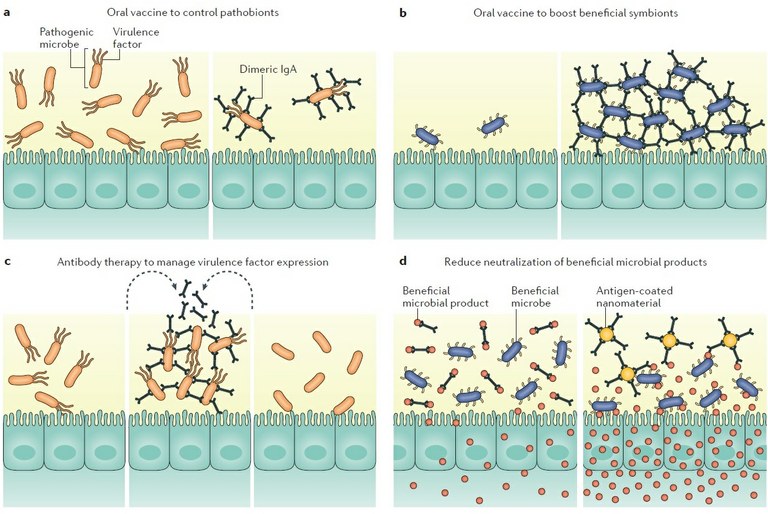
Figure 2: Potential immunotherapies to modulate antibody-mediated immunoselection in the gut. Manipulating antibody-mediated immunoselection (AMIS) to establish a healthy gut can be achieved by enhancing or limiting antibody responses to desired antigens. Oral vaccination could be used to specifically limit pathobiont expansion (part a) or to promote biofilm formation by beneficial species (part b). Exogenous antigens could be delivered into the gut to manage virulence factor expression by pathobionts (part c). Sequestration of antibodies against beneficial microbial products could increase their availability to the host or to other beneficial microorganisms (part d).